
.png)
Combination- and Borderline Products: How to Successfully Navigate Regulatory Uncertainty
By Mahsa Aspsäter, Principal Consultant, Epista Life Science
Borderline products sit in regulatory no-man’s-land, in many cases somewhere between medicinal products and medical devices, not to mention cosmetics.
The same is true for drug–device combinations: figuring out which legislation applies and which parts to comply with can be challenging. I’ve seen companies head down the wrong regulatory path, only to restart months later – a mistake that brings costly delays and setbacks.
Here's how to get it right from the start.
Why classification is so hard
Borderline products sit right between regulatory frameworks. Despite clear definitions in regulations, real-life scenarios can be far from straightforward. The key question always comes down to: Is this a medicinal product or a medical device?
In parallel, drug–device combinations are on the rise, think drug administration devices, implants incorporating a drug, or even a dental filling with antibiotics.
Here, knowing exactly which regulation and which parts to comply with, can be tricky.
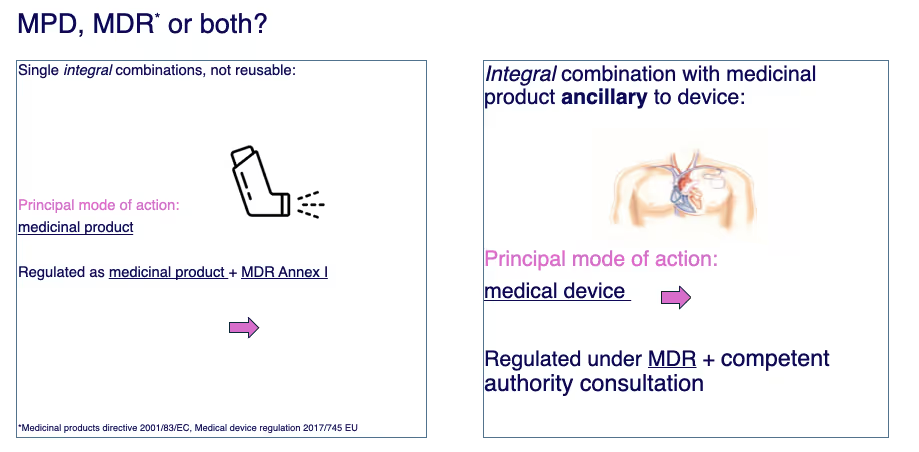
Principal Mode of Action
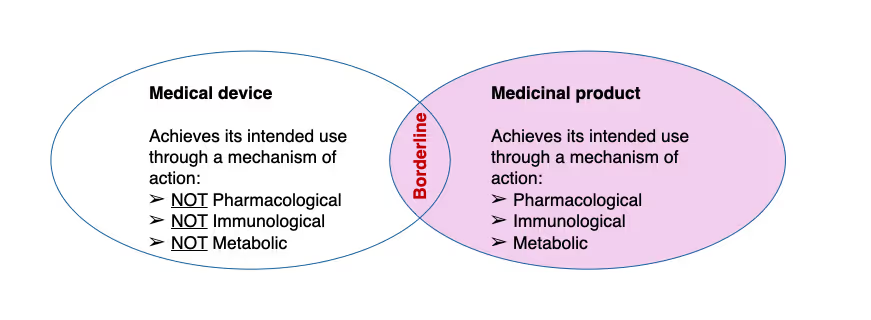
This is where everything starts: determining the "Principal Mode of Action" (PMOA). Simply put, it's identifying the primary mechanism by which your product achieves its intended use and whether that action is achieved by pharmacological, immunological, or metabolic (PIM) means (if so, it is a medicinal product; if not, it may be a device).
In combination products, the PMOA of the product helps you navigate the regulatory pathway and know which part(s) of the respective regulations to comply with.
Let me give you two examples of combination products that show how this works in practice:
The asthma inhaler: Its primary purpose is clearly medicinal – delivering a drug to treat asthma symptoms. This makes it regulated primarily as a medicinal product.
But here's the catch: for an integral drug–device combination authorized as a medicinal product, the marketing authorisation dossier must include evidence that the device constituent conforms to the relevant MDR Annex I General Safety and Performance Requirements. If the device is not already CE‑marked for that specific intended purpose, a Notified Body opinion is required, per Article 117. (This is not required for “plain” Class I devices; it is required for Class Is/Im and above).
The pacemaker lead with antibiotic coating: Here, the medical device (pacemaker lead) provides the principal action, and the antibiotic plays a supportive role. Such products are regulated under MDR but the Notified Body must seek a scientific opinion on the ancillary medicinal substance from a competent authority or the EMA. EMA consultation is mandatory only for substances derived from human blood or plasma.
As a manufacturer, you will need to provide quality, safety, and clinical usefulness data for the ancillary substance as used in the device. Quality information is often structured in CTD Module 3 format; and relevant non-clinical/clinical data are included as applicable. The Notified Body forwards the dossier to the consulted authority.
In more complex combination products, the challenge isn't just identifying the PMOA – it's building a case that regulators will accept.
PMOA determination becomes especially complex when dealing with substance-based borderline medical devices.
The tricky world of substance-based medical devices
Here's where things get really complex.
When you walk into a pharmacy, you'll find many self-care substance-based medical devices sitting right next to OTC medicinal products. They look the same, feel the same, and often have similar indications.
Take salicylic acid – you can find it in both medicinal products and substance-based medical devices. What distinguishes them?
If a product achieves its intended use through pharmacological, immunological, or metabolic action, it's a medicinal product. If not, it may qualify as a medical device.
But here's what's changed: Under MDR, substance-based medical devices face much stricter scrutiny. The new Rule 21 up‑classifies devices composed of substances that are introduced via a body orifice or applied to the skin and are absorbed or locally dispersed to class IIa/IIb/III depending on local versus systemic action and risk. Rule 14 may also apply to a substance-based device, which directly leads to a class III medical device.
The grey area is vast, and I can tell you from experience – regulators are increasingly skeptical about what seemed acceptable under the old Medical Device Directive.
Building your case under MDR
The regulatory bar for medical devices has risen significantly. Regulators now expect precise, evidence-based justifications – vague arguments no longer pass review.
Two documents can be your lifeline here:
- MDCG 2022-5 guidance – This has been developed over many years by EU Commission working groups. It details what pharmacological action actually means, what immunological action means, and all the definitions you really need to understand.
- The borderline and classification manual – This contains past classification cases and decisions. Think of it as regulatory case law. The products aren't named, but you'll see information about which ingredients they contained and what they were intended for. Don’t forget the Helsinki Procedure, which is the process authorities use to resolve borderline/classification disputes.
I can't tell you how many times I've read through these documents. You learn something new every time, depending on which product you're working with.
When disputes arise
Even with solid documentation, disputes happen. When notified bodies start asking questions, you need robust evidence supporting your classification rationale.
If you can't reach agreement, cases escalate to consultations with competent authorities, and occasionally even to court proceedings. I've seen this process play out many times, and having the right regulatory expertise from the start makes all the difference.
Your strategy for success
Borderline products are tricky – there's no getting around that. And MDR has made them much tougher to navigate. But here's what works:
- Define your PMOA clearly with solid scientific rationale
- Choose an experienced notified body – their borderline expertise prevents costly delays
- Get expert regulatory guidance early rather than trying to figure it out alone
One more thing to keep in mind: some EU member states are more skeptical than others. Understanding your target markets is crucial for planning your approach, and engaging early with the relevant competent authority (e.g., the Swedish MPA) can de‑risk divergent interpretations.
The regulatory landscape for borderline products will only get more complex as technology advances. Getting your classification strategy right from the start isn't just about compliance – it's about competitive advantage.
Need a second opinion on your borderline product?
At Epista, we specialize in guiding companies through the regulatory complexities of borderline and combination products. If you're facing classification challenges or preparing for a regulatory submission, let's connect and find the right path forward.